Sun Nov 23 16:31:01 PST 2008
Computerized Crystal Gazing
I was impressed to read that a significant improvement in the ability of computational methods to compute the structure of crystals has been made. The innovation comes from Marcus Neumann and Marc Perrin who show how first principles density functional theory (DFT) calculations can be combined with empirical attraction potentials to rank hypothetical crystal models accurately on the basis of calculated energies.
For molecular crystals this methodology has significantly improved the predictions which are obtained from computational methods.
For inorganic systems, where dispersion forces are less important, it has long been possible to throw the constituent atoms into a suitable box, and use energy minimization to obtain a prediction of the energy of the system. The image below provides and example of such a prediction for the rather simple case of TiO2 in its rutile form. The text in white beneath each structure shows the energy of the system, which declines in successive models, to the final energy, around 223eV, in the final structure, which is shown bottom right.
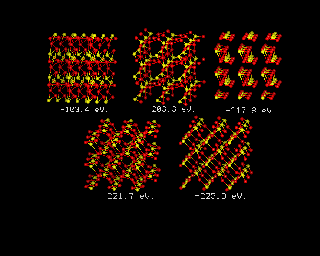
For both molecular crystals and inorganic systems, the theoretical methods can be improved still further, for example: It is possible to include temperature effects, either directly through the use of molecular dynamics, or using a harmonic model. The accuracy of binding energies can be improved. The speed of calculations can be increased.
However, as Neumann and Perrin, and their coworkers, Kendrick and Leusen have shown, computational techniques are now capable of revealing the details of molecular crystal packing in advance of experiment. The results of the computations will not be borne out on every occasion. However, the technology is advancing, and will continue to advance.